

Research
We think big, about very, very small.
Challenges and Multi-Scale Modeling in Solid-State Li-Ion and Mg Batteriesj
Breathtaking colors of our planet
Button
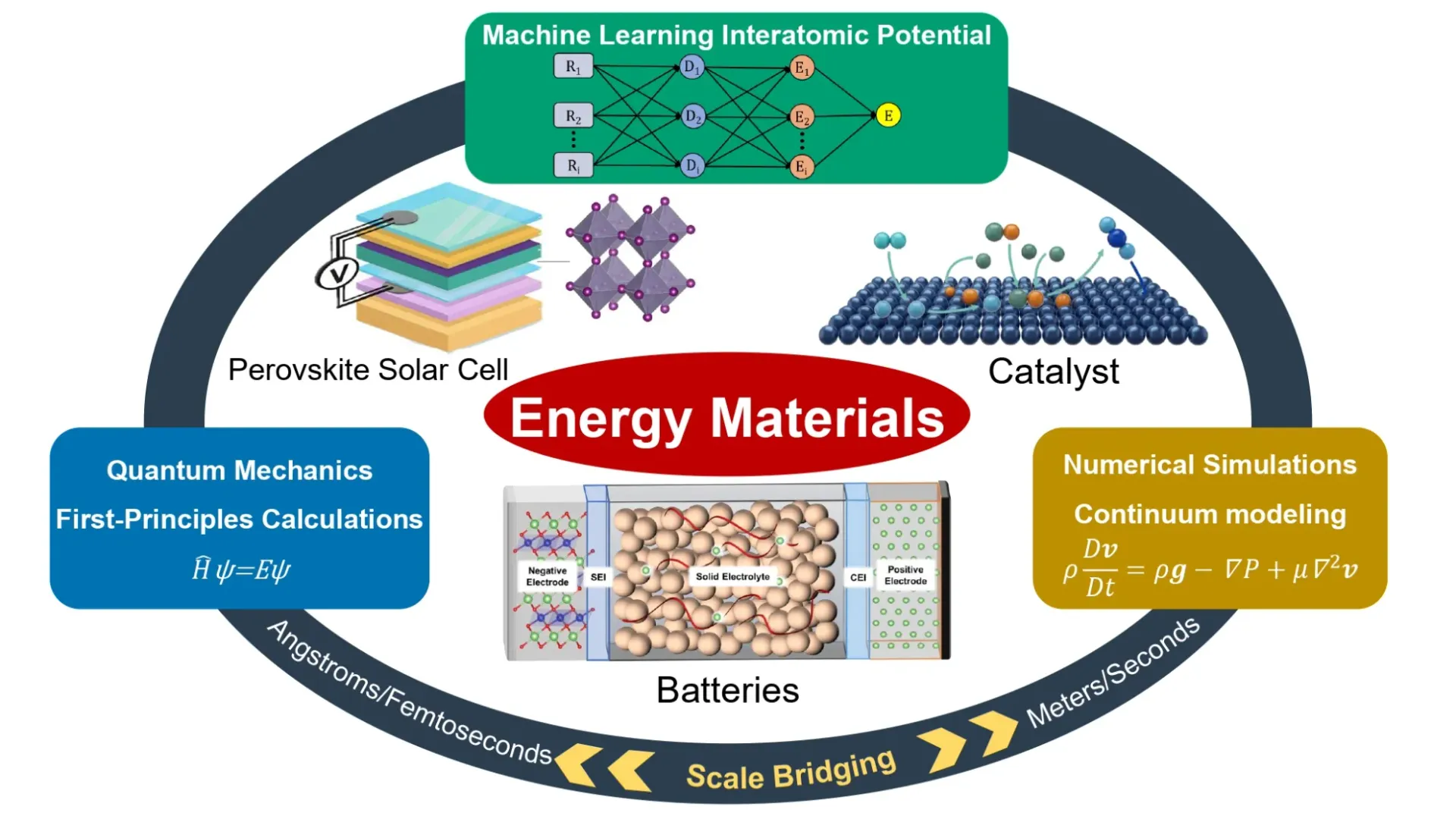
We study energy materials across batteries, catalysts, and perovskite solar cells. We investigate how transport, reactions, and degradation emerge in the bulk, across boundaries and interfaces, and at the device scale. By linking quantum mechanics, machine-learning interatomic potentials, and continuum modeling, we bridge scales from Å/fs to m/s.
Focus areas:
- Batteries (all-solid-state Li-ion, Na-ion, and Mg): ionic transport and conductivity, interphase chemistry, dendrite initiation/suppression, and chemo-mechanical reliability.
- Catalysis: electrocatalysis, photocatalysis, and photoelectrocatalysis with their activity descriptors, reaction pathways, charge/ion transfer, stability under operating conditions, and lifetime prediction.
- Perovskite solar cells: defect chemistry, ion migration, interfacial energetics, and degradation mechanisms.
How we work (multiscale & scale-bridging)
- First-Principle Calculations: atomic-scale structures, energetics, charge transfer, diffusion barriers, and reaction mechanisms.
- Machine-Learning Interatomic Potentials (MLIP): near-DFT/AIMD accuracy for large-system, long-timescale simulations; energy-landscape exploration for configuration prediction.
- Finite Element Analysis (FEA): coupled reactive transport, electrochemical kinetics, heat transfer, and electro-chemo-mechanics at device-relevant scales.
We aim to uncover performance and degradation mechanisms and translate them into practical design rules, including guiding materials selection, device architecture, and durability engineering for novel energy technologies.
For more details, please refer to the Publication section.